


摘 要:运用分子动力学方法,采用Hamid模型构建初始结构,模拟了水化硅酸钙的结构,并分析比较了不同状态水化硅酸钙结构的异同,且通过与试验数据的比较验证了模拟结果的可靠性。模拟结果表明:晶态水化硅酸钙的结构长、短程都有序,而其无定形态只存在近程有序的结构特点;无定形水化硅酸钙的基本结构单元同其晶态一样仍为硅氧四面体,且硅氧四面体链以Q2形式连接;模拟得到的原子间距离、配位数等结构参数基本与实验值相符合。
关键词:水化硅酸钙;结构;分子动力学
0前言
水化硅酸钙是决定水泥性能的关键组分,因此其组成、结构及性质自上世纪50年代Grudemo[1]和Taylor[2]的开创性工作以来一直是水泥科学研究中的重要内容,各国学者都进行了深入研究,提出了一系列结构模型,如类Tobermorite和Jennite模型[3]、固溶体模型[4]、固溶体模型[5]和纳米结构、中介结构假说[6]。同时,水化硅酸钙结构的研究方法和手段也变得越来越丰富,如运用X-射线衍射根据其弥散的衍射峰位置确定其结构大致类似于托贝莫来石[3],运用核磁共振测定水化硅酸钙结构中硅氧四面体的聚合状态[7],运用X-射线光电子能谱根据O1s结合能的变化来区别桥氧与非桥氧[8],运用场发射高分辨透射电镜观察无定形水化硅酸钙中的纳米结晶区[6]。尤其是近年来分子动力学(molecular dynamics, MD)的发展,为在原子水平上探讨水化硅酸钙的精细结构提供了一种崭新的方法。上世纪90年代中期,Faucon[9]就运用分子动力学方法成功地模拟了水化硅酸钙结构不稳定地原因、硅氧四面体链的断裂机理和阳离子(Al)取代硅后对结构的影响,但紧随其后未见任何水化硅酸钙的分子动力学模拟成果见诸于报道。值得注意的是,对用作模型参照的晶态托贝莫来石、羟基硅钙石结构也从晶体学角度上详细地研究了它们的对称性、晶胞参数、键长等
结构性质[10,11],这为构建C–S–H分子动力学模拟的初始结构提供了精确的数据依据。模拟过程中,所使用的分子动力学软件为DL_POLY(http://www.cse.clrc.ac.uk/msi/software/DL_POLY),本课题组对其定义势函数的源代码部分进行了适当的修改。
1 模拟实验
1.1 初始结构创建
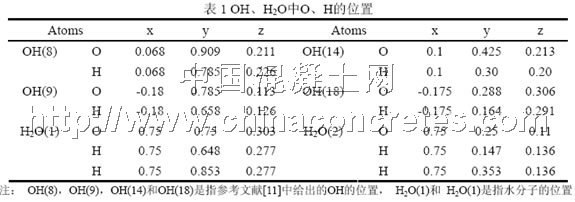
经反复试验本文选择了Hamid模型[11]来构建初始结构。其晶体学参数如下所示。化学式:Ca4Si6O14(OH)4·2H2O;晶系:单斜;晶胞参数:am=0.669 nm,bm=0.739 nm,cm=2.277 nm,γ=123.49°;空间点群:P21;模拟尺寸设为5am×4bm×cm,即am方向尺寸为3.695 nm,bm方向尺寸为2.956 nm,cm方向尺寸为6.831 nm;各原子对应的离子电荷采用经验电荷(formal charges),如Si的电荷取+4。为了考虑Si–H,O–H,H–H的两体作用势和Si–O–H,H–O–H的三体作用势,必须将OH和H2O拆为单个原子。查得O–H键长0.96 nm,∠Si–O–H为109.5°,∠H–O–H为104.5°,于是本文在设定O的位置就是OH/H2O的位置的前提下并根据键长键角求得H的位置,如表1所示。这样处理可能存在如下问题:一是不一定符合原始的对称关系;二是这样生硬地将H放在这一位置可能使之与另一原子相距很近,进而导致排斥强烈,使结构不稳定。因此,为了消除这样处理对结构造成的潜在不利影响,在选定相互作用势及参数后本文还对初始结构进行了优化处理,处理方法如下:在正则系综(NVT)中,300 K下平衡5000步(时间步长为0.1 fs);然后以NVT系综的输出作为微正则系综(NVE)的初始输入,在相同温度下以相同步长再平衡5000步。
1.2 相互作用势设定
该过程的目的就是选择适当的相互作用势经验函数及其参数,并设定分子动力学模拟控制参数。其方法为以本文提出的初始结构作为输入,选定势函数及设定参数后在NVE下运行1000步,其中平衡900步。因为NVE系综在合适的初始条件下能够很快达到平衡(通常为数百步),若系统能够达到平衡且数据不溢出(没有太高的能量作用),则势函数和参数是合理的。
(1)两体作用势经验函数选择及参数设定
由于DL_POLY单独定义库仑势,因此两体作用势就是van de Waals力短程作用势。经过反复试验,本文最终选择了BHM(Born-Huggins-Meyer)势,其函数表达式如式(1),各参数见表2。
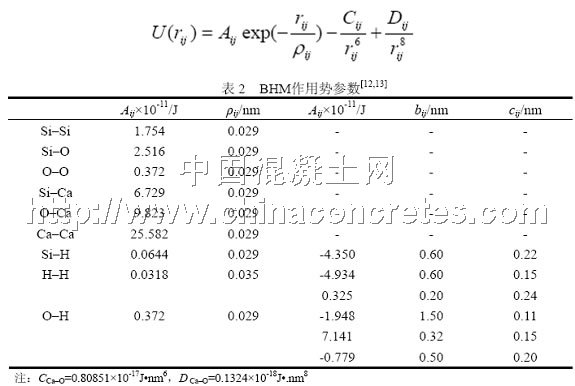
为了体现水的作用(他们有可能离解为H+和OH-),Si–H,H–H,O–H的两体作用势除了BHM势还需加上如式(2)所示的额外项[12,13]。Si–H只需加一项,而H–H则需加两项,O–H更是需加三项,各参数见表2。两体作用势的截断半径取0.76 nm。
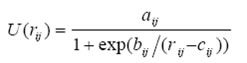
(2)三体作用势经验函数选择及参数设定
三体作用势是一种比van de Waals力作用势更短程、更弱的作用势,其对总能量的贡献通常弱于1%。本文所选三体作用势经验函数的表达式如式(3),其参数如表3所示,截断半径取0.345 nm。

(3)库仑势算法选取及参数设定
根据本文选择的平行六面体周期性边界条件,相应地库仑势算法选用Ewald求和算法。DL_POLY在指定长程作用力(静电作用力)截断和求和精度(相对误差)的前提下,可由其内部函数估算出求和精确性的控制参数,包括收敛参数α,静电作用力在正空间的截断半径rcut和最大倒易矢量。
1.3 结构模拟
采用NVE系综,速度和位置算法采用LF(Leap-Frog)算法,边界条件选为平行六面体周期性边界条件,步长设为1 fs,速度、位置、力等轨迹数据和结构能量、总能量、温度等统计数据每10步存取一次,总共平衡20 000步。为了节省模拟时间,还采用了复合时间步(multiple timestep)算法[14]。
2 模拟结果与讨论
2.1 偏径向分布函数
图1为Ca-O,Si-O,O-O,Si-Si的偏径向分布函数(partial radial distribution function, RDF),图中Crystal对应的结构为优化处理过的初始结构,Liquid对应的结构为以优化过的初始结构为输入制备得到的高温结构,Amorphism对应的结构为由高温结构冷却得到的常温结构。
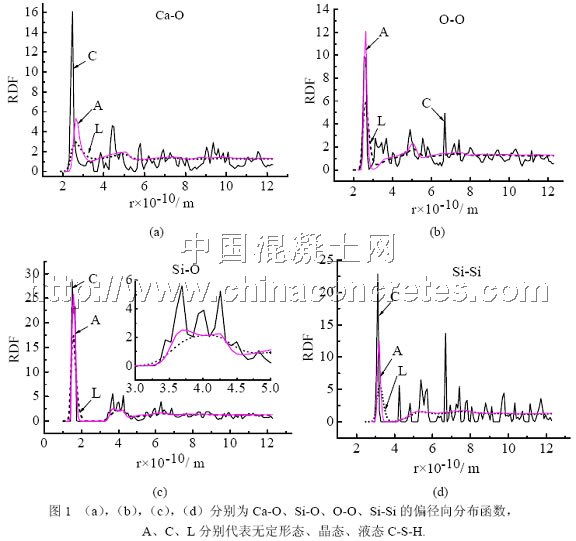
由图1知,Crystal对应结构的各RDF的各层次峰布满整个半径r取值范围,且在较大半径r范围内其值并不趋于1,说明该结构不但近程有序而且远程也是有序的;Amorphism和Liquid对应结构的各RDF只存在较明显的第一近邻峰和明显宽化的第二近邻峰,其他各峰均消失,且在较大半径r范围内其值趋于1,表明Amorphism和Liquid对应结构处于近程有序、远程无序的结构状态。比较Amorphism和Liquid对应结构的各RDF还发现,前者的第一近邻峰都要比后者的尖锐,即前者峰的半高宽要比后者的小,表明前者的近程有序程度要比后者的明显。另外仔细研究Amorphism的Si-O的RDF发现,其第一峰不仅比Crystal的低而且还宽,另外在其第二近邻峰中的4 Å附近出现4了峰的劈裂现象(如图1(c)中的小图所示),这是明显的无定形态特征。综上所述,Crystal对应的结构就是晶态的C-S-H,而Liquid对应的结构则是其高温液态相,Amorphism对应的结构则是无定形的C-S-H。
2.2 配位数
配位数(coordination numbers, CN)在一定程度上也能反应模拟对象的局域结构。比较图2可知,晶态C-S-H对应的各CN曲线在半径r取值范围内总存在着几个明显的台阶,说明它不但有确切的最近邻配位,而且还有次近邻甚至更远近邻的配位,这与晶体的近、远程都有序的结构特征是一致的。液态结构和无定形结构对应的CN曲线只是在较小半径r范围内才有台阶,而在较大范围内曲线几乎呈直线上升,说明他们只存在相距很近的近邻配位,而在相距较远的范围内原子个数与相距半径r几乎呈正比,即不存在特定的长程配位关系。这与液体和无定形体的近程有序而远程无序的结构特点是一致的。
为了进一步考察三者的近程结构,由图2(c)中的小图知,无论是晶体还是无定形体、液体都在CN为4处出现了较宽范围内的长台阶,表明Si的O最近邻配位在三者中都是4配位,说明三者的基本结构单元均为硅氧四面体。另外由图2(d)中的小图知,无定形体在CN等于2处出现了较宽范围内的长台阶且在较大半径r范围内并无其他台阶出现,表明无定形水化硅酸钙中Si与Si之间存在2配位关系,即硅氧四面体链以Q2形式连接,这与Wieker的NMR测试结果非常一致。
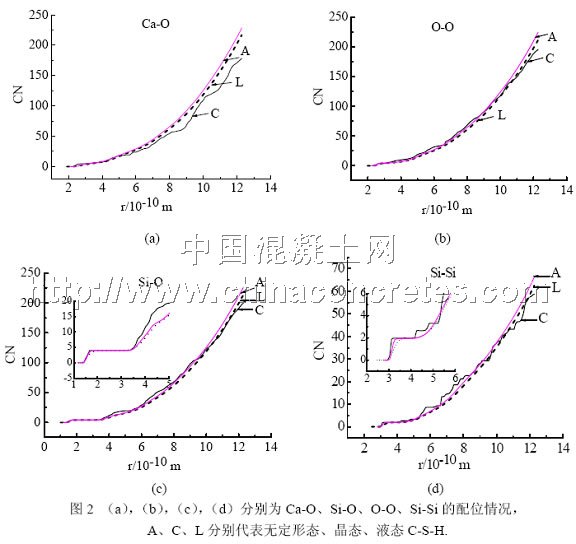
2.3 结构参数
表4为晶态C-S-H和无定形C-S-H的键长或原子间距离、配位数等结构信息。键长或原子间距离取RDF第一近邻峰的峰值对应的半径r。配位数取CN曲线的第一拐点对应的值。
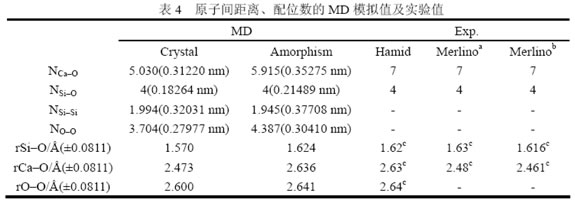
注:圆括号中的数字为计算配位数时的截断半径;a为从文献[14]中得到的0.9 nm托贝莫来石数据;b为从文献[14]中得到的1.1 nm托贝莫来石数据;c为键长平均值。
由表4知,除了Ca的O配位数外其他数据都与实验值基本符合,因此在考虑到模拟体系尺寸有限、边界效应和统计误差的影响的前提下,可以认为本文采用的相互作用势函数和初始结构是合理的,模拟结果基本能重现实际的微观结构。至于Ca的O配位数与实验值有较大差别,其原因在于MD和实验的统计方法不同。
3 结 论
以Hamid的托贝莫来石结构模型为基础,将H2O、OH分解为单个原子按照键长、键角要求置于某一位置,从而构建得到初始结构。由MD模拟得到的偏径向分布函数知,晶态C–S–H不仅长程有序而且短程也有序,而无定形C–S–H确实只存在近程有序的结构特点。由各原子对的配位情况知无定形C-S-H的基本结构单元同晶态C-S-H一样仍为硅氧四面体,且硅氧四面体链以Q2形式连接。在采用合理的相互作用势和初始结构的前提下,MD模拟得到了与实验值基本一致的原子间距离、配位数等结构参数。
参考文献
[1] A. Grudemo. Discussion of the structure of cement hydration compounds[A]. Proceeding of the third international symposium on the chemistry of cement [C]. London: 1952. 247–253.
[2] H. F. W. Taylor. Proposed structure for calcium silicate hydrates gel [J]. J Am Ceram Soc, 1986, 69(6): 464–467.
[3] H. F. W. Taylor. Cement Chemistry [M]. 2nd edition, London: Thomas telford, 1997. 128–134.
[4] M. W. Grutzeck, T. J. Larosa, S. Kwan. Characteristics of C-S-H gels [A]. Proceeding of the 10th ICCC [C], Gothenburg, Sweden: 1997. 2ii067.
[5] I. G. Richardson, G. W. Groves. The composition and structure of C-S-H in hardened slag cement pastes [A]. Proceedings of the 10th ICCC [C], Gothenburg, Sweden, 1997. 2ii068.
[6] D. Viehland, J. F. Li, L. J. Yuan, et al. Mesostructure of calcium silicate hydrate (C-S-H) gels in portland cement paste: Short-range ordering, nanocrystallinity, and local compositional order [J]. J Am Ceram Soc, 1996, 79(7): 1 731–1 744.
[7] X. Cong, R. J. Kirkpatrick. 29Si MAS NMR study of the structure of calcium silicate hydrate [J]. Adv Cem Based Mater, 1996, 3(3): 144–156
[8] L. Black, A. Stumm, K. Garbev, et al. X-ray photoelectron spectroscopy of aluminium-substituted tobermorite [J]. Cem Concr Res, 2005, 34(1): 51–55
[9] P. Faucon, J. M. Delaye, J Yirlet, et al. Study of the structural properties of the C-S-H (I) by molecular dynamics simulation [J]. Cement and Concrete Research, 1997, 27(10): 1 581–1 590.
[10] S. Merlino, E. Bonaccoris, T. Armbruster. Tobermorites: their real structure and order-disorder (OD) character [J]. American Mineralogist, 1999, 84: 1 613–1 621.
[11] S. a. Hamid. The crystal structure of the 11 Å natural tobermorite Ca2.25[Si3O7.5(OH)1.5].1H2O [J]. Zeitschrift für Kristallographie, 1981, 154: 189–198.
[12] B. P. Feuston, S. H. Garofalini. Oligomerization in silica sols [J]. J Phys Chem, 1990, 94: 5 351–5 356.
[13] F. H. Stillinger, A. Rahman. Revised centred force potentials for water [J]. J Chem Phys, 1978, 68(2): 666–670.
[14] W. Smith, T. R. Forester, I. L. Todorov, et al. The DL_POLY user manual. http://www.cse.clrc.ac.uk/msi/software/DL_POLY














